Introduction
Imagine your blood cells which is normally smooth, round, and flexible, suddenly turning rigid and crescent-shaped, jamming blood vessels and starving your tissues of oxygen. That’s exactly what happens in sickle cell disease (SCD), a serious inherited blood disorder that affects millions of people around the world. And yet, despite its prevalence, it remains widely misunderstood outside the communities it most directly affects.
According to the World Health Organization (WHO), approximately 300,000 babies are born with sickle cell disease every year globally, with the majority in sub-Saharan Africa, India, and the Middle East. In the United States, the CDC estimates around 100,000 Americans are living with SCD — the majority of them Black or African American, though the condition also affects people of Hispanic, South Asian, Mediterranean, and Middle Eastern heritage.
This post covers everything you need to know about sickle cell disease: what it is, what causes it, how it’s diagnosed, what living with it looks like, and — excitingly — how the treatment landscape has been transformed by recent breakthroughs that offer real hope for patients and families.
What Is Sickle Cell Disease?
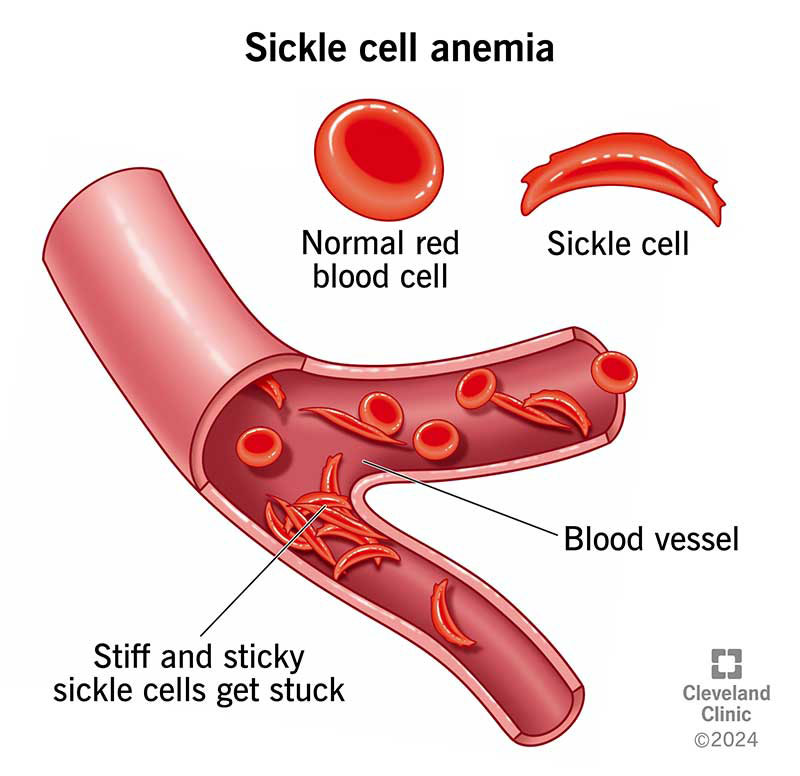
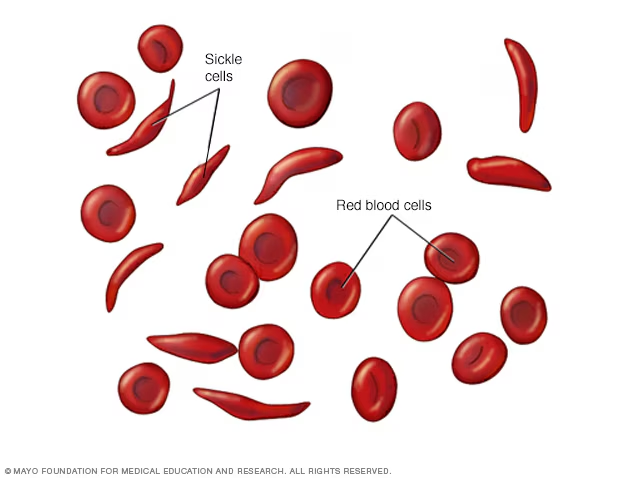
Sickle cell disease is a group of inherited red blood cell disorders caused by a mutation in the gene that produces hemoglobin — the protein inside red blood cells responsible for carrying oxygen through the body. Learn more about how red blood cells and hemoglobin work.
In a healthy person, red blood cells are round, flexible discs that move easily through blood vessels. In someone with SCD, abnormal hemoglobin (called hemoglobin S or HbS) causes red blood cells to warp into a rigid, sickle (crescent) shape — particularly when oxygen levels in the blood are low.
These sickled cells cause two major problems:
- They block blood vessels, cutting off oxygen supply to tissues and organs — triggering sudden, severe pain episodes called vaso-occlusive crises (VOCs)
- They break down rapidly — normal red blood cells live 90–120 days, but sickled cells survive only 10–20 days, leading to chronic anemia. For a deeper look at anemia and what it means for your body, see our post on understanding anemia.
SCD is not one single condition. The most common and severe form is HbSS disease (often called sickle cell anemia), caused by inheriting two copies of the HbS gene — one from each parent. Other variants include HbSC disease and HbS beta-thalassemia, which tend to be somewhat milder but still serious.
What Causes Sickle Cell Disease?
Sickle cell disease is caused by a point mutation in the HBB gene — a single-letter change in the genetic code that tells the body to produce hemoglobin. This mutation results in the production of abnormal hemoglobin S rather than normal hemoglobin A.
How SCD Is Inherited
SCD follows an autosomal recessive inheritance pattern, meaning a child must inherit one defective HBB gene from each parent to develop the disease.
Here’s how it works:
- If both parents carry one copy of the HbS gene (called sickle cell trait), there is a 25% chance with each pregnancy that the child will have SCD, a 50% chance the child will have sickle cell trait (a carrier), and a 25% chance the child will have no sickle cell genes at all.
- People with sickle cell trait (one HbS gene, one normal gene) generally do not have SCD but can pass the gene on to their children.
Why Is SCD More Common in Certain Populations?
The geographic distribution of SCD is no accident. Carrying the sickle cell trait provides some protection against severe malaria — a disease historically prevalent in tropical and subtropical regions. This evolutionary advantage meant that the HbS gene was naturally selected for in populations in sub-Saharan Africa, the Mediterranean, the Middle East, and parts of India. Over generations and through migration, the gene spread worldwide.
This is one of medicine’s most compelling examples of a genetic trait that is both a survival advantage in one context and a cause of serious disease in another.
Signs and Symptoms of Sickle Cell Disease
SCD can affect virtually every organ system in the body. Symptoms typically appear in early childhood — often as young as 5 to 6 months of age — when fetal hemoglobin (which protects newborns) begins to be replaced by adult hemoglobin.
Pain Crises (Vaso-Occlusive Crises)
The hallmark of SCD is the pain crisis — sudden episodes of severe pain that occur when sickled cells block blood flow to bones, joints, the chest, or the abdomen. These crises can be triggered by:
- Cold temperatures or sudden temperature changes
- Dehydration
- Physical or emotional stress
- Infections
- High altitude or low-oxygen environments (including airplane travel)
Pain crises vary widely in severity and duration — from a few hours to several weeks — and are the most common reason for emergency room visits and hospitalizations in people with SCD.
Chronic Anemia
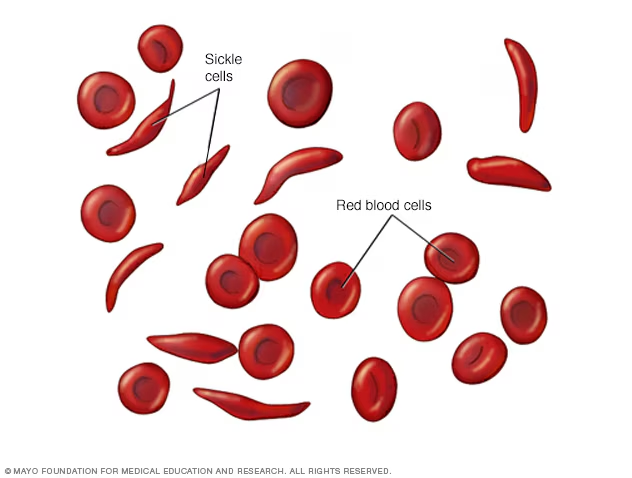
Because sickled red blood cells break down so quickly, people with SCD live with persistent hemolytic anemia. Symptoms include ongoing fatigue, pallor, jaundice (yellowing of the skin and eyes), and shortness of breath. See our post on recognizing signs of anemia for a full breakdown.
Acute Chest Syndrome
Acute chest syndrome (ACS) is a serious, potentially life-threatening complication involving inflammation and sickling in the blood vessels of the lungs. It presents with chest pain, fever, and difficulty breathing and requires urgent hospital treatment.
Stroke
Children with SCD are at significantly elevated risk of stroke — sickled cells can block the major arteries supplying the brain. The NIH reports that without preventive treatment, up to 11% of children with HbSS disease will have a stroke before age 20.
Other Complications
Long-term complications include:
- Splenic sequestration — the spleen traps large numbers of blood cells, causing dangerous drops in red blood cell levels (especially in young children)
- Avascular necrosis — loss of blood supply to bone tissue, commonly affecting the hip
- Priapism — prolonged, painful erections caused by blood flow obstruction (affects males)
- Leg ulcers
- Pulmonary hypertension (high blood pressure in the lungs)
- Kidney damage
- Eye complications (sickle cell retinopathy)
How Is Sickle Cell Disease Diagnosed?
Newborn Screening
In most countries with established healthcare systems, SCD is detected at birth through newborn screening programs. In the United States, all 50 states screen for SCD as part of the standard newborn blood spot test. Early detection is critical — starting preventive care before symptoms appear dramatically improves outcomes.
Hemoglobin Electrophoresis
The definitive diagnostic test is hemoglobin electrophoresis (or HPLC — high-performance liquid chromatography), which separates different types of hemoglobin in a blood sample and identifies the presence and proportion of HbS. This test can diagnose SCD and sickle cell trait at any age.
Prenatal Testing
For families with known carrier status, prenatal diagnosis is available via chorionic villus sampling (CVS) at 10–13 weeks of pregnancy, or amniocentesis at 15–20 weeks. Preimplantation genetic testing (PGT) is also available for couples undergoing IVF.
If you’re unsure of your carrier status, genetic counseling and a simple blood test can provide clarity. See our guide to genetic testing and family planning for more information.
Treatment Options: From Daily Management to Potential Cures
For decades, treatment for SCD focused on managing symptoms rather than addressing the root cause. That has changed significantly in recent years.
Preventive and Supportive Care
Foundational management includes:
- Penicillin prophylaxis — Daily low-dose penicillin in young children (typically from 2 months to 5 years) to prevent life-threatening bacterial infections, especially pneumonia caused by Streptococcus pneumoniae
- Vaccinations — People with SCD need all routine vaccines plus additional ones (pneumococcal, meningococcal, influenza) due to functional asplenia (reduced spleen function)
- Folic acid supplementation — Supports red blood cell production
- Hydration — Dehydration is a key trigger for pain crises; staying well-hydrated is essential
- Regular monitoring — Routine checks of kidney function, eye health, blood pressure, and transcranial Doppler ultrasound in children (to screen for stroke risk)
Hydroxyurea
Hydroxyurea (also called hydroxycarbamide) remains the most widely used disease-modifying medication for SCD. It works by increasing the production of fetal hemoglobin (HbF) — a form of hemoglobin that doesn’t sickle — which dilutes the effects of HbS. Clinical evidence consistently shows it reduces the frequency of pain crises, acute chest syndrome episodes, and hospitalizations, and may improve long-term survival. It is now recommended for most people with HbSS disease from infancy onward, per 2023 guidelines from the American Society of Hematology (ASH).
Blood Transfusions
Regular blood transfusions reduce the proportion of sickled cells in circulation and are used preventively in children at high risk of stroke (identified by transcranial Doppler screening) and in the management of severe complications. Chronic transfusion therapy carries its own risks, including iron overload, which requires treatment with iron chelation therapy.
Voxelotor and Crizanlizumab
Two newer medications have been approved in recent years:
- Voxelotor (Oxbryta) — Approved by the FDA in 2019 and expanded in 2021, it works by increasing hemoglobin’s affinity for oxygen, preventing red blood cells from sickling. Note: As of 2024, the manufacturer voluntarily withdrew voxelotor from the market pending further trial data; patients currently on the drug should consult their hematologist.
- Crizanlizumab (Adakveo) — Approved in 2019, it reduces the ability of sickled cells to stick to blood vessel walls, helping prevent pain crises. Ongoing studies continue to evaluate its long-term effectiveness.
This is an area where the evidence is still developing, and treatment decisions should always be made with a specialist in SCD.
Gene Therapy: A Historic Breakthrough
The most significant recent development in SCD treatment is the arrival of gene therapy. In December 2023, the U.S. FDA approved two gene therapies for SCD:
- Casgevy (exagamglogene autotemcel) — The first approved therapy to use CRISPR-Cas9 gene editing technology, developed by Vertex Pharmaceuticals and CRISPR Therapeutics. It edits the patient’s own stem cells to reactivate fetal hemoglobin production.
- Lyfgenia (lovotibeglogene autotemcel) — Developed by bluebird bio, it uses a viral vector to insert a functional copy of the hemoglobin gene into the patient’s stem cells.
Both therapies are one-time treatments. Early clinical trial data has shown dramatic reductions in pain crises — with many patients remaining crisis-free for years after treatment. These therapies represent the closest thing to a cure currently available, though they are currently accessible only at specialized centers and carry significant cost and procedural complexity.
Bone Marrow (Stem Cell) Transplant
A hematopoietic stem cell transplant (HSCT) from a matched donor can cure SCD by replacing the patient’s bone marrow with healthy marrow that produces normal hemoglobin. It is currently most effective in children and requires a matched sibling donor, limiting its availability. It carries significant risks, including graft failure and graft-versus-host disease.
Living with Sickle Cell Disease: Daily Life and Self-Management
SCD is a lifelong condition, but with the right support and management strategies, many people with SCD lead full, active lives.
Avoiding Crisis Triggers
Being aware of your personal triggers and taking steps to minimize exposure makes a real difference:
- Stay hydrated — Aim for consistent fluid intake throughout the day, especially in hot weather or during exercise
- Stay warm — Cold is a common trigger; dress in layers and avoid prolonged cold exposure
- Manage stress — Emotional stress can trigger crises; mindfulness, therapy, and adequate sleep support overall resilience. See our post on stress management strategies for practical guidance
- Avoid overexertion — Physical activity is encouraged but should be moderate; listen to your body
- Don’t smoke — Smoking worsens vascular damage and oxygen delivery. See why smoking is particularly dangerous with blood disorders
Mental Health and Psychosocial Support
Living with a chronic, painful condition carries a heavy psychological burden. Depression and anxiety are significantly more prevalent in people with SCD than in the general population, according to a 2022 review in Blood Advances. Access to mental health support, peer support groups, and patient advocacy organizations is an essential — and often under-resourced — part of comprehensive SCD care.
Organizations like the Sickle Cell Disease Association of America (SCDAA) and the American Sickle Cell Anemia Association (ASCAA) provide community resources, advocacy support, and educational materials for patients and families.
SCD in Pregnancy
Pregnancy in women with SCD requires specialized care. SCD increases the risk of maternal complications (including pain crises, preeclampsia, and blood clots) and fetal complications (including preterm birth and low birth weight). With careful monitoring by a multidisciplinary team — including a hematologist and maternal-fetal medicine specialist — many women with SCD have healthy pregnancies. See our post on managing chronic illness in pregnancy for further guidance.
Key Takeaways
- Sickle cell disease is an inherited blood disorder caused by a mutation in the hemoglobin gene, causing red blood cells to become rigid and crescent-shaped — blocking blood flow and breaking down rapidly.
- SCD is most prevalent in people of African, Mediterranean, Middle Eastern, and South Asian descent, with approximately 300,000 babies born with SCD each year worldwide (WHO).
- The condition causes a wide range of complications including pain crises, chronic anemia, stroke, acute chest syndrome, and organ damage.
- Treatment has evolved rapidly — hydroxyurea remains the cornerstone disease-modifying drug, and two FDA-approved gene therapies (Casgevy and Lyfgenia) approved in December 2023 represent a historic step toward a functional cure.
- Daily self-management — staying hydrated, avoiding triggers, maintaining regular medical care, and prioritizing mental health — significantly improves quality of life for people living with SCD.
Closing: Hope Is Real — and So Is the Need for Action
Sickle cell disease is no longer a condition for which medicine has little to offer. From refined supportive care protocols to truly transformative gene therapies, the outlook for people diagnosed with SCD today is meaningfully better than it was even a decade ago. Research is accelerating, advocacy is growing, and the voices of the SCD community are rightly becoming louder in shaping healthcare policy and research priorities.
If you or someone you love is living with SCD, the most important steps are to establish care with a hematologist experienced in SCD, stay current with new treatments as they emerge, and connect with the patient community for support. And if you don’t know your sickle cell carrier status — particularly if you have family roots in regions where SCD is prevalent — a simple blood test can give you information that matters deeply for your family’s future.
Explore more on ChiidHealth.com