NF1-PN causes debilitating tumor growth. Explore the causes, life-altering impact, and the difficult fight for treatment in this comprehensive guide to a rare genetic disorder.
Introduction
For individuals and families touched by neurofibromatosis type 1 (NF1), a common genetic condition affecting about 1 in 2,500 to 1 in 3,000 people worldwide, life is often marked by an invisible, internal struggle. Among its most challenging manifestations are plexiform neurofibromas (PN)—complex, often disfiguring, and sometimes painful tumors that grow along nerves. NF1-PN represents a profound medical puzzle: a benign tumor that behaves in a far-from-benign way, causing significant morbidity and altering lives.
For decades, the treatment landscape was bleak, limited to repeated, often disfiguring surgeries with high rates of recurrence. Today, that landscape is shifting. The advent of targeted drug therapies offers new hope, yet significant challenges in diagnosis, management, and access persist. This article delves into the biology of NF1-PN, explores the physical and emotional toll it takes, and examines the difficult road toward effective treatment that balances hope with harsh reality.
Understanding the Foundation: What Are NF1 and Plexiform Neurofibromas?
NF1 is an autosomal dominant genetic disorder caused by a mutation in the NF1 gene on chromosome 17. This gene is responsible for producing a protein called neurofibromin, a critical tumor suppressor that acts as a “brake” on cell growth, primarily by regulating the RAS signaling pathway. When one copy of the gene is mutated from birth, cells are left with only half the normal amount of this crucial protein, a state known as haploinsufficiency. This sets the stage for the varied symptoms of NF1.
Plexiform neurofibromas develop when a Schwann cell (a cell that forms the protective sheath around nerves) loses its one remaining functional NF1 gene. This “second hit” completely removes the growth brake, allowing a tumor to form. Unlike common cutaneous neurofibromas, PNs are deep-seated, involving multiple nerve fascicles and branches, often giving them a “bag of worms” texture. They are believed to be congenital or develop very early in life, with up to 50-60% of individuals with NF1 having at least one PN.
Recognizing the Signs: Diagnosis and the Spectrum of Impact
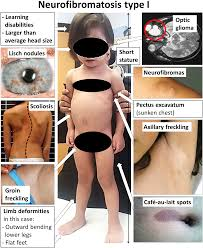
Diagnosis of NF1 is primarily clinical, based on established criteria. A key criterion is the presence of two or more neurofibromas of any type, or one plexiform neurofibroma. Other signs include six or more café-au-lait spots (light brown birthmarks), freckling in skin folds, Lisch nodules in the eyes, and specific bone abnormalities.
| Diagnostic Clue | What It Is | Why It Matters for PN |
|---|---|---|
| Plexiform Neurofibroma | A tumor involving multiple nerve bundles, often felt as a soft, ropy mass under the skin. | Its presence alone fulfills one major diagnostic criterion for NF1. |
| Café-au-Lait Spots | Flat, light brown skin patches present from birth or early childhood. | Having ≥6 spots is a primary indicator, prompting closer examination for PN. |
| Axillary/Groin Freckling | Clusters of small freckles in skin folds (Crowe’s sign). | A common feature that supports the NF1 diagnosis, raising suspicion for internal tumors. |
| Family History | A first-degree relative (parent, sibling, child) with diagnosed NF1. | Allows for proactive monitoring and early imaging to detect asymptomatic PN. |
The impact of PN is as varied as the tumors themselves. Many are asymptomatic, discovered incidentally. Others cause severe morbidity due to their location, size, or rate of growth. Symptoms can include:
- Pain: Chronic, often neuropathic pain is a frequent and debilitating complaint.
- Disfigurement: PNs, especially in the head, neck, or limbs, can cause significant visible distortion and social stigma.
- Functional Impairment: Tumors can compress nerves, leading to weakness, numbness, loss of function, or difficulties with ambulation.
- Airway or Organ Compression: Life-threatening complications can arise from tumors pressing on the airway, spine, or major blood vessels.
The Looming Shadow: The Risk of Malignant Transformation
One of the most serious challenges in managing NF1-PN is the risk of malignant transformation into a malignant peripheral nerve sheath tumor (MPNST), an aggressive and often deadly cancer. MPNSTs are the leading cause of cancer-related mortality in the NF1 population.
The lifetime risk for an individual with NF1 to develop an MPNST is estimated between 8% and 15.8%. These cancers often arise from pre-existing plexiform neurofibromas, sometimes progressing through a premalignant stage known as an atypical neurofibromatous neoplasm of uncertain biologic potential (ANNUBP).
This dire risk underscores the critical need for vigilant, lifelong surveillance. A sudden change in a PN—such as rapid growth, a new onset of severe pain, or neurological deficit—is a red flag requiring immediate medical evaluation.
Navigating the Maze: Treatment Challenges and Emerging Strategies
Historically, surgery was the only option for PN, but it is fraught with challenges. Because PNs are intricately entwined with nerves, complete removal is often impossible without causing severe neurological damage. Subtotal resection is common, but tumors frequently regrow. The decision to operate requires careful weighing of potential benefits against the risks of permanent disability.
A Paradigm Shift: The Era of MEK Inhibitors
The last five years have witnessed a revolutionary change with the approval of MEK inhibitors, the first effective medical therapy for inoperable, symptomatic PN.
- How They Work: These drugs (like selumetinib and mirdametinib) target the overactive RAS/MAPK signaling pathway caused by the lack of neurofibromin. By blocking the MEK protein, they can shrink tumors or halt their growth.
- The Impact: Clinical trials have shown these medications can lead to significant tumor volume reduction (often defined as ≥20% shrinkage) and, importantly, meaningful reductions in pain and improvements in quality of life and physical function.
- The Reality: Treatment is not a cure. It is often lifelong, and side effects like rash, diarrhea, and fatigue are common. Furthermore, not all patients respond, and access can be limited by cost and healthcare system hurdles.
The table below outlines the current central pillars of managing symptomatic NF1-PN:
| Treatment Modality | Key Purpose | Major Considerations & Challenges |
|---|---|---|
| Active Surveillance | Monitor asymptomatic or stable PN for growth or concerning changes. | Requires regular clinical exams and MRIs. Anxiety of “watchful waiting.” Crucial for early cancer detection. |
| Surgery | Debulk or remove tumors causing imminent danger, severe pain, or functional loss. | High risk of nerve damage and recurrence. Often cannot achieve complete resection. |
| MEK Inhibitor Therapy | Shrink or stabilize inoperable, symptomatic PN to alleviate pain and improve function. | Chronic daily medication with side effects. Not all patients respond. High cost and access barriers. |
| Multidisciplinary Supportive Care | Manage pain, provide physical/occupational therapy, and offer psychological support. | Essential for holistic quality of life. Often requires coordinating care across multiple specialists. |
The Critical Role of Multidisciplinary Care and the Path Forward
Given the multi-system nature of NF1, effective management cannot rest with a single specialist. Optimal care requires a coordinated multidisciplinary team (MDT) that may include:
- Geneticists for diagnosis and family counseling.
- Neurologists/Neuro-oncologists for overall management.
- Surgeons (neuro, orthopedic, plastic) for procedural interventions.
- Oncologists to manage medical therapy and MPNST.
- Pain specialists, physical therapists, and psychologists to support quality of life.
The future holds promise. Research continues on next-generation MEK inhibitors and combination therapies. Active surveillance protocols using advanced whole-body MRI are proving to be lifesaving, allowing for the early detection and removal of premalignant lesions (ANNUBP), thereby preventing progression to MPNST. Advocacy for broader access to effective therapies and specialized care centers remains a paramount challenge for the NF community.
Frequently Asked Questions (FAQs)
Q: Is NF1-PN cancerous?
A: Plexiform neurofibromas themselves are benign (non-cancerous) tumors. However, they carry a risk of transforming into a highly aggressive cancer called a malignant peripheral nerve sheath tumor (MPNST). This is why careful, lifelong monitoring is essential.
Q: Are MEK inhibitors like selumetinib a cure for NF1-PN?
A: No, they are not a cure. MEK inhibitors are a chronic treatment that can effectively shrink tumors and manage symptoms, but they must be taken continuously. If the medication is stopped, tumors typically regrow. They represent a revolutionary management tool, not a definitive solution.
Q: My child was just diagnosed with NF1. Should they get a full-body MRI to look for PN?
A: This is a critical discussion to have with a specialized NF clinic. While not every child needs immediate imaging, a baseline MRI is often recommended, particularly in late adolescence or when transitioning to adult care. The decision is based on clinical symptoms, family history, and the presence of surface findings. A specialized team can best advise on the appropriate timing and modality for surveillance.
Conclusion: A Journey of Resilience and Science
Living with or caring for someone with NF1-PN is a profound journey marked by uncertainty, pain, and resilience. The landscape, however, is changing. From an era defined solely by the surgeon’s knife, we have entered a new age of targeted medicine and proactive surveillance. The challenges—diagnostic delays, treatment side effects, cancer risk, and the emotional burden—remain immense.
Yet, the trajectory points toward greater hope. Through continued research, robust multidisciplinary care, and empowered patient advocacy, the goal is to transform NF1-PN from a debilitating disorder into a manageable chronic condition. It is a fight not just against tumors, but for a future where the promise of science translates into a better quality of life for every individual and family on this difficult path.