Unravel groundbreaking advances in prion disease treatment. From CHARM technology to ION717 clinical trials, explore revolutionary approaches offering hope for Creutzfeldt-Jakob disease and fatal brain disorders affecting families worldwide.
Introduction
Imagine watching a loved one’s personality dissolve before your eyes within months; memories vanishing, coordination failing, cognition collapsing at lightning speed. This nightmare scenario defines prion diseases, among the most devastating neurological conditions known to medicine. These rare disorders affect approximately 350 people annually in the United States, with about 70% dying within one year of diagnosis. Yet amid this darkness, groundbreaking scientific advances are finally illuminating pathways toward treatment and prevention.
Prion diseases represent a unique category of neurodegenerative disorders caused not by bacteria, viruses, or parasites, but by misfolded proteins called prions. These rogue proteins trigger a catastrophic chain reaction in the brain, converting normal proteins into toxic versions that accumulate relentlessly. The result? Rapid brain destruction that leaves families shattered and medicine humbled until now.
Understanding the Prion Protein Nightmare
What Makes Prions Uniquely Terrifying
Prion diseases are caused by infectious abnormal folding of proteins, where misfolded proteins trigger normally folded proteins to also become misfolded. This self-perpetuating cascade distinguishes prions from conventional pathogens. Unlike bacteria or viruses that can be targeted with antibiotics or antivirals, prions resist standard sterilization procedures and persist in tissue long after death.
When examined under a microscope, affected brains appear filled with holes, resembling sponges, hence the medical term transmissible spongiform encephalopathies. These microscopic cavities represent the graveyards of neurons destroyed by toxic prion accumulation.
The Devastating Speed of Neurodegeneration
What makes prion diseases particularly cruel is their velocity. The median duration of illness ranges between 13 and 14 months, though symptoms often worsen within weeks to months of onset. Patients may function normally one month, then require complete care the next. This rapidity leaves families scrambling for answers during the narrow window when loved ones remain conscious.
Types of Prion Diseases
Prion diseases manifest in several forms, each with distinct characteristics:
Sporadic Creutzfeldt-Jakob Disease (sCJD): Approximately 85% of cases occur spontaneously for unknown reasons, striking seemingly at random without identifiable risk factors. These cases represent “totally random bad luck,” occurring stochastically in the brain for reasons scientists still cannot explain.

Genetic Prion Diseases: About 7.5% of cases are inherited in an autosomal dominant manner, passed from parent to child through mutations in the PRNP gene encoding prion protein. These include fatal familial insomnia, Gerstmann-Sträussler-Scheinker syndrome, and familial CJD.
Acquired Prion Diseases: Transmitted through exposure to infected tissue, these historically occurred through contaminated medical instruments, growth hormone derived from cadavers, or dura mater transplants. Variant CJD (vCJD) resulted from consuming beef from cattle infected with bovine spongiform encephalopathy, commonly called “mad cow disease”.
The Clinical Picture: Recognizing Prion Disease
Early Warning Signs
In early stages, patients may develop nonspecific symptoms including vertigo, headache, fatigue, and sleep disorders, followed by memory problems, agitation, irritability, depression, apathy, mood swings, and visual loss. These initial symptoms often lead to misdiagnosis as depression, anxiety disorders, or early Alzheimer’s disease.
Patients with variant CJD often present initially with psychiatric symptoms, behavioral changes, and painful dysesthesias, with movement disorders developing early but dementia appearing as a late sign. This atypical presentation in younger individuals further complicates early recognition.
Progressive Neurological Decline
As disease advances, the clinical picture becomes unmistakable. Symptoms worsen quickly, usually within several weeks to a few months, including personality changes, memory loss, impaired thinking, blurry vision or blindness, problems with coordination, trouble speaking and swallowing, and sudden jerky movements.
The main symptoms include severe mental deterioration and dementia alongside involuntary muscle jerks called myoclonus. Patients may appear startled by ordinary stimuli, develop muscle rigidity, and lose ability to perform basic self-care tasks.
Terminal Stage Complications
Death usually occurs within a year from medical issues associated with the disease, including trouble swallowing, falls, heart issues, lung failure, or pneumonia and other infections. As brain function deteriorates, patients lose consciousness, enter vegetative states, and ultimately succumb to complications of immobility and neurological failure.
Diagnostic Challenges and Modern Testing
The Diagnostic Dilemma
Rapid symptom progression represents one of the most important clues that a person may have prion disease, yet this speed simultaneously complicates definitive diagnosis. Many conditions cause rapidly progressive dementia, requiring systematic evaluation to distinguish prion disease from treatable alternatives.
Advanced Diagnostic Technologies
Brain MRI Imaging: Brain MRI with diffusion-weighted imaging or FLAIR sequences demonstrates sensitivity of 98% and specificity of 93%, typically showing hyperintensities within the basal ganglia, thalamus, and cortex. These characteristic patterns help clinicians recognize prion disease even before confirmatory testing.
Cerebrospinal Fluid Analysis: CSF testing examines elevations in 14-3-3 and tau proteins, non-specific markers of rapid brain cell death seen in CJD. While these proteins indicate neuronal destruction, they lack specificity for prion disease.
RT-QuIC Testing: Real-time quaking-induced conversion assay represents a much more specific test for prion disease, detecting the prions that cause CJD. This revolutionary diagnostic tool can identify minuscule amounts of misfolded prion protein in cerebrospinal fluid, enabling diagnosis during life with unprecedented accuracy.
Electroencephalography: EEG recordings may reveal characteristic periodic sharp wave complexes in certain CJD subtypes, though this finding occurs inconsistently.
Genetic Testing: For suspected familial cases, sequencing the PRNP gene identifies disease-causing mutations, enabling predictive testing for at-risk family members.
Confirmatory Diagnosis
Brain biopsy or examination of brain tissue after death represents the gold standard to confirm prion disease presence, but clinicians can often establish accurate diagnoses before death through comprehensive clinical assessment combined with modern testing technologies.
The Historic Failure of Treatment Attempts
Decades of Disappointment
Several compounds including flupirtine, quinacrine, pentosan polysulfate, and doxycycline have been used on a trial basis for patients with prion disease, yet none demonstrated significant benefit in rigorous studies.
Doxycycline Trials: A multi-institutional, double-blind randomized trial of 121 CJD patients treated with doxycycline versus placebo showed no significant difference in survival or disease progression. Researchers speculated that oral treatment after symptom onset arrives too late to alter disease trajectory.
Pentosan Polysulfate: Some patients received this compound through experimental protocols, with one notable case surviving 10 years after diagnosis—an extraordinary duration. However, systematic trials failed to replicate these anecdotal successes.
Antibody Approaches: The first in-body treatment of six CJD patients with humanized monoclonal antibody PRN100 reported that the antibody could access the brain without clinically significant adverse effects, though all patients showed progressive neurological decline.
Why Traditional Approaches Failed
Most therapeutic candidates targeted prions after brain damage had already occurred. By the time symptoms emerge, massive neuronal loss has already transpired. Additionally, many compounds failed to cross the blood-brain barrier in therapeutic concentrations or demonstrated unacceptable toxicity at effective doses.
Revolutionary Breakthroughs: The Dawn of Hope
CHARM Technology: Silencing Disease at Its Source

Researchers from the Whitehead Institute and Broad Institute developed CHARM—Coupled Histone tail for Autoinhibition Release of Methyltransferase—a set of molecular tools delivered to the brain that adds chemical tags to the prion gene to prevent protein production by cells.
Unlike gene editing that permanently alters DNA sequences, CHARM performs epigenetic editing where chemical tags get added to DNA to turn off or silence target genes without modifying the underlying DNA itself, with the gene remaining intact but switched off permanently. This represents a one-time treatment rather than requiring continuous medication.
Dramatic Results in Preclinical Studies: Research showed that the CHARM system, delivered through a single intravenous injection, could be distributed across the brain in mice and eliminate more than 80% of prion protein. This unprecedented level of protein suppression occurred throughout the brain, not just in localized regions.
The Science Behind CHARM: Previous research demonstrated that mice completely lacking the gene for prion protein resist infectious prions, as prion protein isn’t essential for survival in healthy adults. CHARM exploits this biological reality, silencing prion protein production to prevent the substrate necessary for disease propagation.
ION717: From Laboratory to Clinical Reality
Vallabh and Minikel collaborated with biotech company Ionis Pharmaceuticals to develop ION717, with early clinical trials announced as fully enrolled in December 2024.
Antisense Oligonucleotide Mechanism: ION717 aims to lower prion protein levels by reducing messenger RNA and preventing protein translation through antisense oligonucleotides—short strands of DNA, RNA, or analogs that bind to target genes to modulate expression.
ASOs can reach the brain when delivered at high doses via cerebrospinal fluid injection every few months, requiring repeated administration but avoiding the complexities of gene therapy delivery systems.
Historic First Patient: University Hospitals Cleveland Medical Center and Case Western Reserve University achieved a historic milestone, becoming the first site activated in December 2024 and enrolling the world’s first participant the following month in the international, first-in-human study of prion protein-lowering treatment utilizing antisense oligonucleotide.
The Personal Urgency Behind Innovation
Broad Institute Senior Group Leader Sonia Vallabh switched careers and became a researcher after learning she carries a disease-causing version of the prion protein gene, with she and her husband Eric Minikel now running a lab working to develop drugs that can prevent and treat these diseases.
Their deadline isn’t determined by grant cycles or academic timelines but by the genetic time bomb Vallabh carries. In less than two years, Weissman, Vallabh, and their collaborators developed CHARM technology that can turn off disease-causing genes including the prion protein gene, as well as potentially genes coding for many other proteins implicated in neurodegenerative diseases.
Emerging Therapeutic Strategies
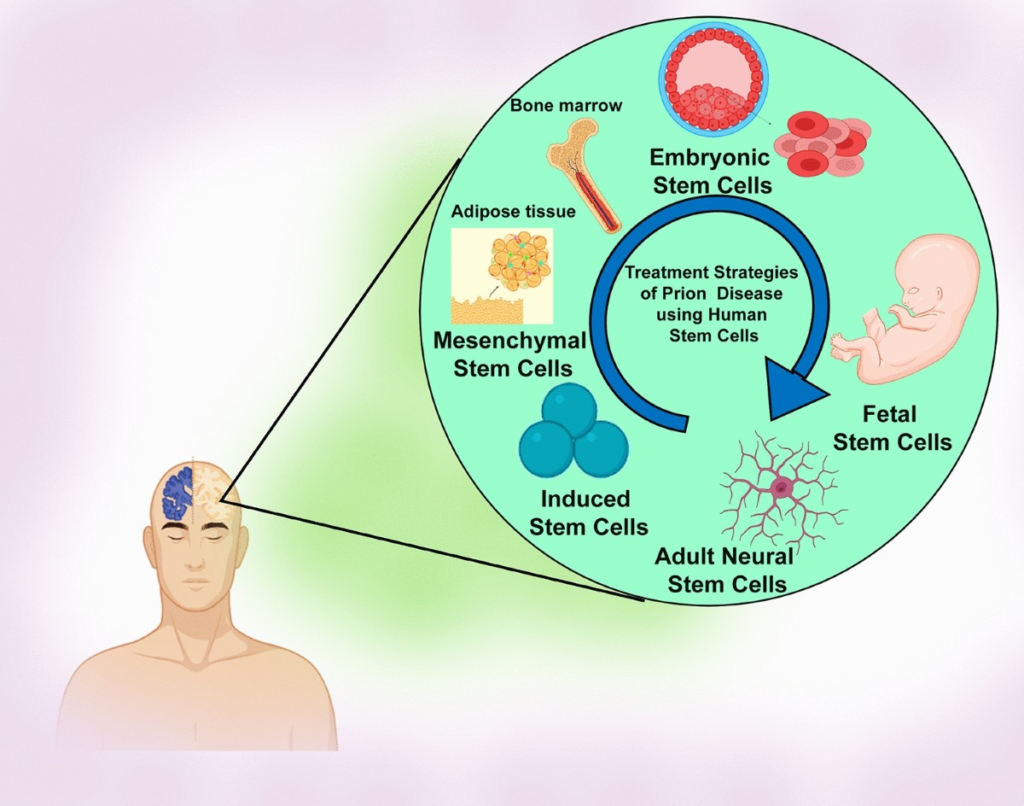
Stem Cell Approaches
Some researchers have addressed stem cell therapies to repair damaged neurons caused by misfolded pathological prions, with the most frequently researched approaches including embryonic stem cells, mesenchymal stem cells, induced pluripotent stem cells, and neuron stem cells.
While stem cells cannot reverse prion accumulation, they might regenerate neural circuits destroyed by disease, potentially extending functional capacity even as underlying pathology continues. This approach shows promise in other neurodegenerative conditions including Parkinson’s disease, Huntington’s disease, and amyotrophic lateral sclerosis.
Immunotherapy Innovations
Current therapeutic strategies have focused on preventing or clearing infectious prion particles, with newer antibody designs targeting both cellular prion protein to prevent conversion and pathogenic prions to facilitate clearance.
Challenges include achieving sufficient brain penetration, avoiding immune-related complications, and intervening early enough to preserve neurological function. The PRN100 experience demonstrated that antibodies can reach brain tissue safely, establishing proof-of-concept for this approach despite disappointing efficacy results.
Targeted Protein Degradation
Novel approaches utilizing nanoformulations aim to synergistically enhance the solubility, permeability, and targeting capability of targeted protein degradation drugs, thereby reducing the risk of off-target toxicity. These systems could selectively eliminate misfolded prions while preserving normal cellular proteins.
Living with Prion Disease: Management and Support
Symptom Management Strategies
Currently there’s no cure for CJD or any other prion disease, with treatments focusing on making the person comfortable and easing symptoms through medications that may help relieve behavioral changes, seizures, and muscle jerks.
Pain Management: Opioid medications can address discomfort as disease progresses, with dosing adjusted to maintain comfort without excessive sedation during remaining conscious periods.
Seizure Control: Antiepileptic medications including valproate and clonazepam help manage involuntary movements and myoclonus, improving quality of life even when disease progression continues.
Psychiatric Symptoms: Antianxiety medications, antidepressants, and sometimes antipsychotics may alleviate behavioral disturbances, agitation, and psychiatric symptoms, particularly in variant CJD presentations.
Nutritional Support
During later stages of disease, people may need IV fluids and machine feeding, though because prion diseases are incurable, these approaches can have limited use. Families face difficult decisions about artificial nutrition and hydration as swallowing difficulties progress and consciousness fades.
Palliative and Hospice Care
Given the inevitably fatal prognosis, early integration of palliative care principles benefits patients and families. Hospice services provide comprehensive support during final weeks, addressing physical comfort, emotional needs, and spiritual concerns while honoring individual preferences.
Family and Caregiver Considerations
The Emotional Toll
Watching prion disease consume a loved one creates profound psychological trauma. The rapidity of decline leaves families in crisis mode, struggling to adapt as capabilities vanish weekly. Unlike slowly progressive dementias where adjustment occurs gradually, prion disease compresses years of decline into months.
Genetic Testing Dilemmas
For familial prion disease cases, family members face agonizing decisions about predictive genetic testing. Learning one carries a disease-causing mutation without available preventive treatment creates existential distress. Conversely, not knowing leaves uncertainty hanging over life planning.
The emerging treatment landscape changes this calculus. With ION717 trials underway and CHARM technology advancing, genetic testing enables early intervention before symptoms appear—potentially preventing disease onset entirely.
Support Resources
Organizations including the CJD Foundation provide education, support networks, and advocacy for affected families. Connecting with others navigating similar devastation offers validation and practical guidance during an otherwise isolating experience.
Prevention and Risk Reduction
Sporadic CJD: Unavoidable Misfortune
Sporadic CJD has no known cause, occurring randomly without identifiable prevention strategies. No lifestyle modifications, dietary changes, or screening tests reduce sporadic CJD risk.
Preventing Acquired Prion Disease
Food Safety: Animal inspections help keep BSE-infected cattle out of the food supply, though uninspected or improperly processed meat can still pose risk. Avoiding brain tissue, spinal cord, and certain other tissues from ruminant animals minimizes theoretical transmission risk.
Medical Procedures: Healthcare facilities implement rigorous sterilization protocols for instruments contacting neural tissue. Single-use equipment for spinal procedures eliminates recontamination risks.
Blood Supply Protection: Blood donation restrictions exclude individuals with familial CJD history, prior dura mater grafts, and human growth hormone derived from cadavers, protecting transfusion recipients.
Chronic Wasting Disease Concerns
Chronic wasting disease affects deer, elk, and moose populations across North America. While no human cases have been definitively linked to CWD, hunters should avoid consuming neural tissue from affected animals and consider testing harvested game in endemic regions.
The Economics of Rare Disease Treatment
Cost Considerations for Novel Therapies
If ION717 proves effective, treatment will likely be costly, as typical for rare disease therapies, with Ionis’s spinal muscular atrophy drug nusinersen having a list price of $750,000 for the first year of injections and $375,000 annually thereafter.
For diseases as rare and devastating as prion disorders, pharmaceutical economics create challenging dynamics. Development costs remain substantial regardless of patient population size, while limited markets constrain revenue potential. These factors drive high pricing when treatments emerge.
Access and Equity Issues
Expensive treatments raise profound equity concerns. Will life-saving therapies remain accessible only to wealthy individuals or those with exceptional insurance coverage? How will healthcare systems in lower-income countries afford these interventions?
Patient advocacy organizations and policymakers must address these questions proactively, ensuring scientific breakthroughs translate into broadly accessible treatments rather than benefits limited to privileged populations.
Frequently Asked Questions
What exactly is a prion and how does it cause disease?
A prion is a misfolded version of a normal protein found throughout the body, particularly in the nervous system. When prions contact normal prion proteins, they trigger those normal proteins to misfold as well, creating a self-perpetuating chain reaction. These accumulating misfolded proteins form toxic clumps that kill brain cells, leaving the characteristic sponge-like holes visible under microscopy. Unlike bacteria or viruses, prions contain no genetic material and resist standard sterilization methods.
Can prion disease be inherited, and should I get genetic testing?
Yes, approximately 10-15% of prion disease cases result from inherited genetic mutations in the PRNP gene. If you have a first-degree relative diagnosed with prion disease, particularly young-onset cases or multiple affected family members, genetic counseling can clarify your risk. Testing reveals whether you carry disease-causing mutations. With emerging treatments like ION717 entering clinical trials, identifying mutation carriers before symptom onset may enable preventive intervention—a paradigm shift from when testing offered only distressing certainty without treatment options.
How quickly does prion disease progress, and what can patients expect?
Prion diseases progress with devastating rapidity. From symptom onset to death typically spans four to six months, with 90% of patients dying within one year. Initial symptoms may include memory problems, mood changes, or coordination difficulties. Within weeks to months, severe dementia develops alongside involuntary muscle movements, vision loss, and progressive loss of all voluntary functions. Patients ultimately require complete care, lose consciousness, and succumb to complications including infections and organ failure. This compressed timeline distinguishes prion diseases from other dementias that typically progress over years.
Is there any cure or effective treatment for prion diseases currently available?
Currently, no cure or treatment slows prion disease progression in symptomatic patients. However, revolutionary developments offer genuine hope. The ION717 antisense oligonucleotide entered human clinical trials in late 2024, representing the first therapy specifically designed to lower prion protein levels in the brain. Additionally, CHARM technology demonstrated ability to eliminate over 80% of prion protein in preclinical studies through a single intravenous injection. While neither approach has proven effective in humans yet, these represent quantum leaps beyond previous failed attempts.
Can prion disease be transmitted through casual contact or caregiving?
No, prion diseases are not contagious through ordinary social contact. You cannot contract prion disease from touching, hugging, sharing meals, or providing routine care for affected individuals. Transmission requires direct exposure to infected brain or spinal cord tissue through medical procedures, contaminated surgical instruments, or—in the case of variant CJD—consuming infected beef products. Healthcare workers and caregivers face no elevated risk compared to the general population when following standard precautions.
What’s the difference between Creutzfeldt-Jakob disease and Alzheimer’s disease?
While both conditions cause dementia, they differ fundamentally. Alzheimer’s disease results from accumulation of amyloid plaques and tau tangles, progressing gradually over years with initial memory difficulties eventually affecting all cognitive domains. Prion diseases like CJD involve misfolded prion proteins that propagate rapidly, causing symptoms to emerge and worsen within weeks to months rather than years. CJD also typically includes prominent involuntary muscle movements (myoclonus) and coordination problems earlier in the disease course compared to Alzheimer’s. Diagnostic testing including MRI patterns, CSF analysis, and particularly RT-QuIC testing distinguishes these conditions.
Should I avoid certain foods or behaviors to prevent prion disease?
For sporadic CJD the most common form, no known dietary or lifestyle factors influence risk, as cases occur randomly. To minimize theoretical risk of variant CJD from mad cow disease, avoid consuming brain tissue, spinal cord, or neural tissues from cattle, sheep, or other ruminants, particularly from regions with documented BSE outbreaks. Hunters in areas with chronic wasting disease should avoid consuming nervous system tissues from deer, elk, or moose. Beyond these specific precautions, no evidence suggests any foods, activities, or environmental exposures alter prion disease risk.
The Future of Prion Disease Treatment
Nanoformulation Advances
Major efforts to improve pharmacokinetics involve development of nanoformulations incorporating liposomes, cyclodextrins, polymers, and other unique delivery systems, with nanoparticle formulations potentially circumventing poor oral absorption issues and improving deposition into tumor tissue.
These advanced delivery platforms could transport therapeutic agents across the blood-brain barrier more efficiently, enabling lower doses with reduced systemic side effects while maximizing brain tissue concentrations. Nanotechnology may finally solve the drug delivery challenge that has hindered numerous promising compounds.
Combination Therapy Approaches
Future protocols may combine multiple mechanisms: gene silencing to prevent new prion production, antibodies to clear existing pathogenic prions, and neuroprotective agents to preserve remaining neurons. This multi-pronged strategy mirrors successful approaches in other complex diseases like cancer and HIV.
Preventive Treatment Paradigm
The most transformative potential involves treating asymptomatic mutation carriers before neurological damage begins. Researchers developed an improved method to target the prion gene that could lead to treatments preventing prion diseases before symptoms appear.
Imagine a world where individuals learn they carry prion mutations through genetic testing, then receive one-time gene silencing therapy that eliminates disease risk entirely—transforming a death sentence into a manageable genetic variant. This represents the ultimate goal of current research efforts.
Implications Beyond Prion Disease
It has become clear in the past two decades that abnormal proteins propagate in prion-like ways in Alzheimer’s disease, Parkinson’s disease, and other maladies that destroy neurons, with mechanisms being similar and every reason to think that therapies developed for prion disease could work for other neurodegenerative diseases.
Success in prion disease could unlock treatments for far more common conditions affecting millions worldwide. The protein misfolding and propagation mechanisms underlying prion diseases mirror those in Alzheimer’s, Parkinson’s, and ALS, suggesting that conquering prions may provide blueprints for addressing the broader neurodegenerative disease landscape.
Conclusion
For decades, prion diseases represented medicine’s ultimate failure—uniformly fatal conditions progressing with merciless speed, offering patients and families nothing but suffering and loss. Healthcare providers could only deliver devastating diagnoses followed by inadequate comfort measures while watching helplessly as disease consumed their patients’ humanity within months.
That era is ending. The convergence of advanced molecular biology, gene therapy technologies, and the passionate determination of researchers like Vallabh and Minikel is finally cracking the prion code. ION717 trials are enrolling patients right now. CHARM technology is advancing toward human studies. Stem cell approaches and novel antibodies are under development. For the first time in history, realistic pathways toward preventing and treating prion diseases exist.
The journey from breakthrough to broadly available treatment remains long and uncertain. Clinical trials may disappoint. Regulatory hurdles will test patience. Economic challenges will require creative solutions. But the scientific foundation has been laid. The impossible is becoming possible.
For families affected by prion diseases, for individuals carrying genetic mutations, and for the rare but devastating sporadic cases that continue appearing unpredictably, these developments offer something previously unimaginable: genuine hope. The revolution in prion disease treatment has begun, and with it comes the promise that these devastating conditions may finally join the list of once-fatal diseases that medicine has learned to prevent and treat.
The race continues, driven by scientific brilliance, personal urgency, and the determination to ensure that no more families experience the devastation of watching prion disease destroy their loved ones. In research laboratories, clinical trial sites, and pharmaceutical companies worldwide, scientists and clinicians are working to transform prion diseases from inevitably fatal nightmares into preventable, treatable conditions. That future is closer than ever before.