Latest 2025 guide to Huntington Disease: genetics, CAG repeats, symptoms, breakthrough gene therapy results, and comprehensive treatment options available.
Introduction
Huntington Disease (HD) stands as one of the most devastating inherited neurodegenerative disorders, affecting approximately 1 in 10,000 people worldwide. Unlike many neurological conditions with unknown causes, HD’s genetic foundation has been clearly identified, yet this knowledge has paradoxically made the disease both more predictable and more tragic—individuals can know decades in advance that they will develop this progressive, ultimately fatal condition.
Named after Dr. George Huntington, who first described the disease in 1872, HD is characterized by the relentless progression of motor dysfunction, cognitive decline, and psychiatric symptoms that typically appear in midlife. The disease strikes during what should be the most productive years, affecting not only patients but entire family systems for generations.
However, 2024 and 2025 have brought unprecedented hope to the HD community. Revolutionary gene therapy approaches are showing remarkable promise, with recent clinical trials demonstrating a 75% slowing of disease progression, while advances in understanding the underlying biology are opening new therapeutic avenues previously thought impossible.
This comprehensive guide explores the complex genetics underlying HD, the devastating progression of symptoms, the latest breakthrough treatments including gene therapy, and what these developments mean for patients, families, and the future of neurodegenerative disease treatment.
Understanding Huntington Disease: The Basics
Definition and Overview
Huntington Disease is an autosomal dominant inherited disorder caused by a mutation in the huntingtin (HTT) gene located on chromosome 4. This single genetic defect leads to progressive degeneration of specific brain regions, particularly the basal ganglia and cerebral cortex, resulting in the characteristic triad of motor, cognitive, and psychiatric symptoms.
Key Characteristics:
- Inheritance Pattern: Autosomal dominant (50% chance of inheriting from affected parent)
- Age of Onset: Typically 30-50 years, but can range from childhood to late adulthood
- Progression: Relentlessly progressive over 15-20 years
- Prevalence: Affects 3-7 per 100,000 individuals of European ancestry
- Life Expectancy: 15-20 years from symptom onset
The Huntingtin Gene and Protein
The HTT gene produces huntingtin protein, a large protein essential for normal cellular function. In healthy individuals, huntingtin plays crucial roles in:
- Intracellular transport: Moving materials within cells
- Gene transcription: Regulating gene expression
- Cell survival: Protecting against programmed cell death
- Synaptic transmission: Supporting communication between neurons
- Development: Critical for normal brain development
When mutated, the huntingtin protein becomes toxic, gradually destroying the very cells it was meant to protect.
The Genetics of Huntington Disease
CAG Repeat Expansion: The Molecular Basis
The genetic mutation causing HD involves an expansion of CAG (cytosine-adenine-guanine) trinucleotide repeats within the HTT gene. In people with Huntington’s disease, the CAG segment is repeated 36 to more than 120 times. People with 36 to 39 CAG repeats may or may not develop the signs and symptoms of Huntington’s disease, while people with 40 or more repeats almost always develop the disorder.
CAG Repeat Categories:
Normal Range (10-35 repeats):
- No risk of developing HD
- Stable transmission to offspring
- Normal huntingtin protein function
Intermediate Range (36-39 repeats):
- Reduced penetrance—may or may not develop HD
- Risk increases with repeat number
- Potential for expansion in offspring
Full Mutation (40+ repeats):
- If you have 40 copies of the CAG repeats one of your HD genes, you will develop symptoms of HD over the course of a natural lifespan
- Inevitable disease development
- Earlier onset with higher repeat numbers
Juvenile HD (60+ repeats):
- Onset before age 20
- More rapid progression
- Often inherited from affected fathers
Somatic Instability: A Growing Understanding
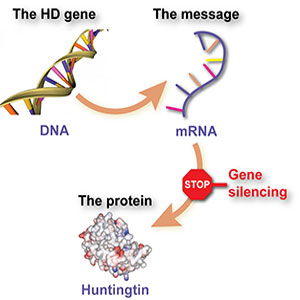
Recent breakthroughs in 2024 and 2025 have revealed that “2024 proved a year where many breakthroughs in our understanding of somatic instability were made.” This phenomenon refers to the continued expansion of CAG repeats throughout an individual’s lifetime, even after birth.

Key Discoveries:
- Somatic CAG expansion occurs throughout life and understanding the impact of somatic expansion on neurodegeneration is key to developing therapeutic targets
- Single-cell measurement of the Huntington’s disease-causing CAG repeat reveals that somatic expansion of this repeat drives pathological changes in neurons
- Different tissues show varying rates of expansion
- Brain regions most affected by HD show highest expansion rates
Clinical Implications:
- Somatic expansion may explain variable disease progression
- Targeting expansion could slow disease progression
- New biomarkers for tracking disease development
Inheritance Patterns and Anticipation
HD follows autosomal dominant inheritance, meaning:
- Each child of an affected parent has a 50% chance of inheriting the mutation
- Both males and females can be affected equally
- The disease can be passed from either parent
- No “skipping” of generations occurs
Genetic Anticipation: The phenomenon where successive generations show earlier onset and more severe symptoms, particularly with paternal transmission. This occurs because:
- Sperm production is more prone to CAG expansion than egg production
- Juvenile HD cases are almost exclusively paternally inherited
- Each generation may have progressively longer CAG repeats
Genetic Testing and Counseling
Predictive Testing: Available for at-risk individuals before symptom onset, but presents profound ethical and psychological challenges:
- Benefits: Life planning, reproductive decisions, clinical trial eligibility
- Risks: Psychological distress, discrimination, impact on family relationships
- Uptake: Only 15-20% of at-risk individuals choose testing
Prenatal Testing:
- Chorionic villus sampling (10-12 weeks)
- Amniocentesis (15-18 weeks)
- Preimplantation genetic diagnosis (PGD)
Testing Process:
- Pre-test genetic counseling
- Psychological assessment
- Blood sample collection
- Laboratory analysis
- Results disclosure with support
- Post-test counseling and follow-up
Clinical Manifestations and Disease Progression
The Classic Triad of Symptoms
HD affects three major domains simultaneously, with symptoms typically emerging in midlife and progressing over 15-20 years:
Motor Symptoms
Chorea: The hallmark movement disorder
- Involuntary, dance-like movements
- Initially subtle, becoming more pronounced
- Affects face, limbs, and trunk
- Worsens with stress and emotion
- May improve with concentration
Progressive Motor Decline:
- Early Stage: Subtle coordination problems, clumsiness
- Middle Stage: Obvious chorea, difficulty with fine motor tasks
- Late Stage: Bradykinesia (slow movement), rigidity, dystonia
Specific Motor Impairments:
- Gait abnormalities and falls
- Speech difficulties (dysarthria)
- Swallowing problems (dysphagia)
- Eye movement abnormalities
- Loss of voluntary motor control
Cognitive Symptoms
Executive Function Deficits: Often the first cognitive changes
- Difficulty with planning and organization
- Impaired decision-making
- Problems with multitasking
- Reduced mental flexibility
Memory Changes:
- Working memory impairment
- Difficulty learning new information
- Preserved long-term memory initially
- Progressive memory decline in later stages
Language and Communication:
- Word-finding difficulties
- Reduced verbal fluency
- Comprehension difficulties in advanced stages
- Communication becomes increasingly challenging
Cognitive Progression:
- Mild: Subtle changes in executive function
- Moderate: Obvious cognitive impairment affecting daily tasks
- Severe: Dementia requiring full-time care
Psychiatric Symptoms
Often the most devastating aspect for families, psychiatric symptoms can precede motor symptoms by years:
Depression: Most common psychiatric symptom
- Affects 40-60% of HD patients
- Can occur 10-15 years before motor symptoms
- Often includes suicidal ideation
- May be reactive or part of the disease process
Irritability and Aggression:
- Explosive outbursts disproportionate to triggers
- Verbal and sometimes physical aggression
- Strains family relationships significantly
- Often responds to behavioral interventions and medications
Anxiety Disorders:
- Generalized anxiety common
- Social anxiety due to embarrassment about symptoms
- Panic attacks in some individuals
- Recent research shows associations between huntingtin gene (HTT) CAG repeat size variations and psychiatric phenotypes
Psychosis:
- Less common but can occur
- Paranoid delusions
- Hallucinations (rare)
- Usually responds to antipsychotic medications
Apathy and Loss of Initiative:
- Reduced motivation and interest
- Social withdrawal
- Decreased self-care
- Often confused with depression
Disease Stages and Functional Decline
Stage 1 (Early/Preclinical):
- Duration: 10-15 years before diagnosis
- Subtle cognitive and psychiatric changes
- Possible minor motor signs
- Functional independence maintained
Stage 2 (Early Symptomatic):
- Duration: 3-5 years
- Obvious movement disorder emerges
- Cognitive decline becomes apparent
- May still work with accommodations
Stage 3 (Middle):
- Duration: 5-10 years
- Significant functional impairment
- Unable to work or drive
- Requires assistance with complex tasks
Stage 4 (Advanced):
- Duration: 5-8 years
- Severe motor, cognitive, and psychiatric symptoms
- Requires assistance with basic daily activities
- High care needs
Stage 5 (End-Stage):
- Duration: 2-5 years
- Profound disability
- Requires total care
- Death usually from complications (pneumonia, falls)
Pathophysiology: How HD Destroys the Brain
Cellular Mechanisms of Toxicity
Mutant Huntingtin Protein (mHtt): The expanded CAG repeats produce an altered huntingtin protein with toxic properties:
- Protein Aggregation: mHtt forms clumps that interfere with cellular function
- Nuclear Inclusion Bodies: Protein aggregates in cell nuclei
- Impaired Protein Clearance: Overwhelmed cellular cleanup systems
- Mitochondrial Dysfunction: Energy production severely compromised
Selective Vulnerability: HD preferentially affects specific brain regions:
- Striatum: Caudate nucleus and putamen most severely affected
- Cortical Areas: Particularly motor and frontal regions
- Medium Spiny Neurons: Specific cell type showing greatest vulnerability
- Neural Circuits: Cortico-striatal-thalamic loops disrupted
Neuroinflammation and Glial Activation
Recent research has found that complement proteins and microglia mediate early and selective loss of corticostriatal synapses, revealing inflammation as a key driver of HD pathology:
Microglial Activation:
- Brain’s immune cells become overactive
- Release of inflammatory substances
- Contributes to neuronal death
- Potential therapeutic target
Astrocyte Dysfunction:
- Support cells lose normal function
- Reduced neuroprotective capacity
- Impaired glutamate uptake
- Contributes to excitotoxicity
Synaptic Dysfunction
Before neurons die, their connections (synapses) become dysfunctional:
- Reduced Synaptic Transmission: Communication between neurons impaired
- Loss of Synaptic Plasticity: Reduced ability to adapt and learn
- Circuit Disruption: Entire brain networks become dysfunctional
- Functional Connectivity Loss: Brain regions can’t communicate effectively
Current Treatment Approaches
Symptomatic Treatments
Motor Symptom Management:
Chorea Treatment:
- Tetrabenazine (Xenazine): First FDA-approved HD treatment
- Reduces chorea by depleting dopamine
- Side effects: depression, parkinsonism, sedation
- Requires careful monitoring
- Deutetrabenazine (Austedo): Newer, improved version
- Longer half-life, less frequent dosing
- Similar efficacy with potentially fewer side effects
- Better tolerated by most patients
Other Motor Medications:
- Antipsychotics: Haloperidol, olanzapine for severe chorea
- Benzodiazepines: For dystonia and muscle spasms
- Baclofen: Muscle relaxant for rigidity
Psychiatric Treatment:
Depression Management:
- SSRIs: First-line antidepressants (sertraline, citalopram)
- Mirtazapine: Helpful for depression with weight loss
- Tricyclic antidepressants: For treatment-resistant cases
- Psychotherapy: CBT and supportive counseling
Behavioral Symptoms:
- Mood stabilizers: Valproic acid, lithium
- Antipsychotics: For aggression and psychosis
- Anxiolytics: Short-term use for severe anxiety
Cognitive Symptoms:
- No FDA-approved medications for HD-related cognitive decline
- Cholinesterase inhibitors: Limited evidence
- Stimulants: May help attention and executive function
- Cognitive rehabilitation: Behavioral interventions
Supportive Care and Rehabilitation
Physical Therapy:
- Maintain mobility and prevent complications
- Balance training to reduce fall risk
- Stretching to prevent contractures
- Adaptive equipment recommendations
Occupational Therapy:
- Modify activities of daily living
- Home safety assessments
- Assistive technology
- Energy conservation techniques
Speech Therapy:
- Communication strategies
- Swallowing safety
- Alternative communication methods
- Family education
Nutritional Support:
- High-calorie diet due to increased energy expenditure
- Texture modifications for dysphagia
- Supplementation as needed
- Feeding tube considerations in late stages
Multidisciplinary Care
HD Care Centers: Specialized clinics providing comprehensive care:
- Neurologist with HD expertise
- Genetic counselor
- Social worker
- Physical/occupational/speech therapists
- Psychiatrist or psychologist
- Nutritionist
Care Coordination:
- Regular comprehensive assessments
- Coordinated treatment planning
- Family support and education
- Community resource connections
- Clinical trial opportunities
Breakthrough Treatments: Gene Therapy and Beyond
Revolutionary Gene Therapy Results
The year 2024 has marked a turning point in HD treatment with unprecedented clinical trial results:
AMT-130 Gene Therapy (uniQure): For patients receiving the high-dose, the data showed a statistically significant 75% slowing of disease progression as measured by cUHDRS (p=0.003). Treated patients had a mean change in cUHDRS from baseline of 0.38 points compared to 1.52 points for matched participants in the control group.
How AMT-130 Works:
- One-time surgical delivery to the brain
- Uses adeno-associated virus (AAV) vector
- Delivers micro-RNA to reduce mutant huntingtin production
- Specifically targets the striatum, most affected brain region
Clinical Trial Results:
- 3-year follow-up data available
- Significant slowing of functional decline
- Safety profile acceptable
- Some patients showing stabilization or improvement
Implications:
- First treatment to significantly modify HD progression
- Potential to transform HD from fatal to manageable condition
- Regulatory approval being sought
- May be available within 2-3 years
Other Gene-Targeted Approaches
Antisense Oligonucleotides (ASOs):
- Short DNA sequences that reduce huntingtin production
- Delivered via lumbar puncture
- Roche’s tominersen showed mixed results in trials
- Newer generations in development
RNA Interference (RNAi):
- Uses cellular machinery to silence huntingtin gene
- Multiple companies developing approaches
- Challenges with delivery to brain
- Promising preclinical results
Allele-Specific Targeting:
- Aim to reduce only mutant huntingtin, preserving normal protein
- More challenging but potentially safer approach
- Several approaches in development
- May avoid potential side effects of total huntingtin reduction
Stem Cell and Regenerative Therapies
Cell Replacement Strategies:
- Replace dead neurons with healthy ones
- Multiple cell types being investigated
- Challenges with integration and function
- Early clinical trials beginning
Neural Progenitor Cells:
- Cells that can develop into neurons
- May promote endogenous repair
- Some clinical trials completed
- Mixed results to date
Induced Pluripotent Stem Cells (iPSCs):
- Patient’s own cells reprogrammed to become neurons
- Reduces immune rejection risk
- Still experimental
- Promising for personalized therapy
Neuroprotective Approaches
Targeting Somatic Expansion: Based on recent discoveries about ongoing CAG expansion:
- Drugs to prevent further repeat expansion
- May slow or halt disease progression
- Multiple compounds in development
- Represents novel therapeutic approach
Anti-Inflammatory Treatments:
- Target brain inflammation component
- Microglial modulators in development
- Complement system inhibitors
- May be combined with other approaches
Metabolic Enhancement:
- Improve cellular energy production
- Target mitochondrial dysfunction
- Creatine, coenzyme Q10, and others tested
- Limited success to date
Biomarkers and Disease Monitoring
Neuroimaging Biomarkers
Structural MRI:
- Striatal atrophy precedes symptoms by years
- Quantitative measurements track progression
- Used in clinical trials as outcome measures
- Whole brain atrophy in later stages
Functional Imaging:
- PET Scans: Show metabolic changes
- fMRI: Reveals altered brain connectivity
- DTI: Demonstrates white matter changes
- Research tools becoming clinical applications
Fluid Biomarkers
Cerebrospinal Fluid (CSF):
- Mutant huntingtin protein measurable
- Neurofilament light chain indicates neuronal damage
- Multiple biomarkers in development
- Require lumbar puncture for collection
Blood-Based Biomarkers:
- Neurofilament light chain in blood
- Mutant huntingtin in blood cells
- Less invasive than CSF
- Improving sensitivity and specificity
Digital Biomarkers
Smartphone and Wearable Technology:
- Motor function assessment via sensors
- Speech pattern analysis
- Cognitive testing through apps
- Continuous monitoring capabilities
Advantages:
- Remote monitoring possible
- Objective, quantitative measures
- Frequent data collection
- Patient-friendly approach
Living with Huntington Disease
Impact on Patients
Physical Challenges:
- Progressive loss of motor control
- Increasing difficulty with daily activities
- Communication becomes increasingly difficult
- Swallowing and eating problems develop
Emotional and Psychological Impact:
- Grief over progressive losses
- Anxiety about future decline
- Depression common
- Loss of independence and identity
Social Challenges:
- Stigma and misunderstanding
- Employment discrimination
- Relationship strain
- Social isolation
Family Impact
Genetic Burden:
- 50% risk for each child
- Decision-making about genetic testing
- Reproductive choices and family planning
- Guilt and anxiety about transmission
Caregiving Demands:
- Progressive increase in care needs
- Physical and emotional strain
- Financial impact
- Multiple family members may be affected
Children and Adolescents:
- Risk of inheriting the gene
- Witnessing parent’s decline
- Premature role reversal
- Educational and social impacts
Support Systems and Resources
Huntington’s Disease Society of America (HDSA):
- Patient and family support services
- Educational resources and materials
- Advocacy and awareness campaigns
- Research funding and coordination
Support Groups:
- Patient support groups
- Family and caregiver support
- Online communities and forums
- Peer counseling programs
Professional Services:
- Genetic counseling
- Social work services
- Mental health counseling
- Legal and financial planning
Research Frontiers and Future Directions
Emerging Therapeutic Targets
Protein Quality Control:
- Enhance cellular cleanup of toxic proteins
- Autophagy enhancers
- Proteasome activators
- Chaperone proteins
Neuronal Connectivity:
- Restore synaptic function
- Enhance neuroplasticity
- Circuit-specific interventions
- Neuromodulation approaches
Epigenetic Modifications:
- Modify gene expression without changing DNA
- Reverse disease-associated changes
- HDAC inhibitors and other compounds
- Potentially reversible interventions
Precision Medicine Approaches
- Genes that influence HD progression
- Personalized treatment based on genetics
- Risk stratification and prognosis
- Targeted interventions
Biomarker-Guided Therapy:
- Monitor treatment response
- Adjust therapy based on biomarkers
- Predictive models for outcomes
- Personalized treatment protocols
Combination Therapies
Multimodal Approaches:
- Combine different mechanisms
- Gene therapy plus neuroprotection
- Anti-inflammatory plus regenerative
- Maximize therapeutic benefit
Sequenced Interventions:
- Different treatments at different stages
- Prevention in presymptomatic phase
- Symptomatic treatment during progression
- Palliative care in late stages
Prevention and Early Intervention
Presymptomatic Intervention
Lifestyle Factors:
- Exercise: May delay onset and slow progression
- Cognitive Stimulation: Mental activity potentially protective
- Social Engagement: Maintaining relationships and activities
- Stress Management: Reducing psychological stress
Pharmacological Prevention:
- Trials in presymptomatic individuals
- Ethical considerations about treatment without symptoms
- Risk-benefit assessments
- Long-term safety monitoring
Family Planning and Prevention
Genetic Counseling:
- Risk assessment and education
- Reproductive option counseling
- Psychosocial support
- Decision-making assistance
Reproductive Options:
- Preimplantation Genetic Diagnosis (PGD): IVF with genetic testing
- Prenatal Testing: Testing during pregnancy
- Donor Gametes: Using donor sperm or eggs
- Adoption: Alternative family building
Exclusion Testing:
- Indirect genetic testing approach
- Determines inheritance without revealing gene status
- Allows reproductive decisions without knowing personal risk
- Complex but available option
Global Perspectives and Healthcare Systems
International Research Collaboration
Global HD Research Networks:
- Enroll-HD: Worldwide observational study
- HD-CSF: Cerebrospinal fluid research network
- Registry studies: Track natural history globally
- Clinical trial networks: Coordinate international trials
Research Funding:
- Government funding agencies
- Private foundations (CHDI, HDSA, HDA)
- Pharmaceutical company investment
- Patient advocacy-driven research
Healthcare Access and Equity
Global Variations in Care:
- Specialist availability varies widely
- Treatment access differs by region
- Healthcare system coverage varies
- Economic barriers to care
Rare Disease Challenges:
- Limited specialist knowledge
- Misdiagnosis common
- Treatment delays
- Lack of specialized services
Regulatory Landscape
Drug Development Challenges:
- Small patient population
- Long disease course
- Ethical considerations
- Regulatory pathway optimization
Accelerated Approval Pathways:
- FDA breakthrough therapy designation
- European Medicines Agency adaptive pathways
- Compassionate use programs
- Post-market surveillance requirements
Economic and Social Impact
Healthcare Costs
Direct Medical Costs:
- Specialist consultations and monitoring
- Medications and treatments
- Hospitalizations and emergency care
- Long-term care facility costs
Indirect Costs:
- Lost productivity and earnings
- Caregiver burden and lost work
- Disability accommodations
- Social services utilization
Lifetime Cost Estimates:
- Average lifetime cost: $4-6 million per patient
- Higher costs due to early onset
- Multiple family members may be affected
- Costs increase with disease progression
Insurance and Coverage Issues
Coverage Challenges:
- Genetic discrimination concerns
- Long-term care coverage limitations
- Experimental treatment exclusions
- International coverage variations
Policy Implications:
- Genetic Information Nondiscrimination Act (GINA)
- Long-term care insurance needs
- Disability benefit qualification
- Healthcare system adaptations needed
Hope and Future Outlook
Reasons for Optimism
Scientific Breakthroughs:
- Gene therapy showing 75% slowing of disease progression
- Understanding of somatic expansion mechanisms
- Multiple therapeutic approaches in development
- International research collaboration
Technology Advances:
- Digital biomarkers and remote monitoring
- Artificial intelligence in drug discovery
- Gene editing technologies (CRISPR)
- Personalized medicine approaches
Community Strength:
- Strong patient advocacy organizations
- Increased research funding
- Growing awareness and understanding
- Supportive communities and networks
Timeline for Major Advances
Near Term (2025-2027):
- Gene therapy approvals likely
- Additional symptomatic treatments
- Better biomarkers for clinical use
- Expanded clinical trial options
Medium Term (2027-2032):
- Combination therapies
- Prevention trials in presymptomatic individuals
- Regenerative medicine applications
- Improved quality of life interventions
Long Term (2032-2040):
- Potential for disease prevention
- Gene editing applications
- Restoration of lost function
- Transformation from fatal to manageable condition
Challenges Remaining
Scientific Challenges:
- Delivering treatments to the brain
- Reversing established neurodegeneration
- Understanding individual variation
- Long-term safety of new treatments
Healthcare System Challenges:
- Training specialists worldwide
- Ensuring equitable access
- Managing costs of new therapies
- Coordinating complex care needs
Social Challenges:
- Reducing stigma and discrimination
- Supporting affected families
- Ethical considerations with genetic testing
- Preparing for potential prevention scenarios
Conclusion: A New Era of Hope
Huntington Disease has long stood as one of medicine’s most challenging conditions—a devastating genetic disorder with no effective treatments and an inevitably fatal outcome. For decades, families lived with the knowledge that if they carried the gene, they would develop the disease, and there was nothing medical science could do to change that trajectory.
Today, we stand at a remarkable inflection point. The recent clinical trial results showing a 75% slowing of disease progression represent more than just promising data—they signal the beginning of a new era where HD may transform from a fatal diagnosis to a manageable condition.
The convergence of multiple scientific advances is creating unprecedented opportunities:
- Gene therapy approaches that can be delivered directly to affected brain regions
- Understanding of somatic expansion mechanisms that drive disease progression
- Advanced biomarkers that can track disease years before symptoms appear
- Neuroprotective strategies that may preserve brain function
- Regenerative approaches that could restore lost capabilities
Perhaps most remarkably, we’re moving toward treatments that don’t just manage symptoms but actually modify the underlying disease process. The prospect of preventing HD in those who carry the gene—once science fiction—is becoming a realistic goal within the next decade.
For the HD community, this progress brings both hope and new challenges. Families must navigate decisions about genetic testing, clinical trial participation, and treatment timing that previous generations never faced. Healthcare systems must prepare for new therapies that require specialized delivery and monitoring. Society must address questions about access, equity, and the ethical implications of preventing a genetic condition.
The journey from George Huntington’s 1872 description of the disease to today’s breakthrough treatments represents 150 years of scientific progress. The pace of discovery is now accelerating exponentially, with advances measured in months rather than decades.
While significant challenges remain, the HD community can face the future with genuine hope. For the first time, we can envision a world where carrying the HD gene doesn’t mean facing an inevitable, devastating decline. The tools to prevent, slow, and eventually reverse this condition are within our grasp.
The transformation of Huntington Disease from an invariably fatal condition to a treatable disorder would represent one of medicine’s greatest triumphs—and offer hope to millions affected by other neurodegenerative diseases. We stand at the threshold of that transformation today.